Grâce aux nouvelles techniques de séquençage d’ADN, le nombre de séquences de génomes disponibles augmente exponentiellement. Ces séquences renferment le programme génétique à l’origine de la formation des êtres vivants. Le décrypter permettrait de mieux comprendre à la fois le fonctionnement des organismes et leur évolution. Pourtant, ce déchiffrage reste une tâche ardue. C’est particulièrement vrai pour la détection des réseaux dits « transcriptionnels » qui relient des protéines essentielles, les facteurs de transcription, aux gènes qu’ils régulent en s’y fixant. Prédire la structure de ces réseaux à partir de l’analyse des séquences génomiques est un enjeu majeur chez tous les organismes mais les sites de fixation des facteurs de transcription sont de petites régions d’ADN variables en séquence et en position ce qui rend leur détection particulièrement difficile.
Un travail effectué chez les plantes dans notre laboratoire montre qu’il est possible de bâtir des modèles capables de prédire de façon précise les sites reconnus dans un génome par un facteur de transcription. Cette étude est focalisée sur le facteur LEAFY, véritable architecte des structures florales chez les plantes à fleurs.
En combinant des données biochimiques et structurales à des méthodes mathématiques et bio-informatiques innovantes, un modèle biophysique a été bâti qui prédit, avec un pouvoir inégalé, les sites reconnus par LEAFY dans le génome. Ces prédictions ont été validées par une analyse à l’échelle génomique de la fixation de LEAFY réalisée
in vivo chez la plante modèle A
rabidopsis thaliana (technique appelée
ChIP-seq). Ce modèle a ensuite été utilisé sur les différents génomes végétaux disponibles pour tester l’existence de la relation entre LEAFY et un gène qu'il régule (
AGAMOUS) responsable du développement des étamines et des pistils (et donc des fruits et graines). Cette relation avait été établie chez
Arabidopsis mais on ne sait pas quand elle est apparue pendant l’évolution. L’application du modèle sur différentes espèces a montré que ce lien existait déjà il y a plus de 100 millions d'années, avant la divergence entre les monocotylédones (les céréales par exemple) et les dicotylédones (comme la rose ou le pétunia). Par ailleurs, cette étude a mis en évidence la grande variabilité de séquence et de position des sites de liaison de LEAFY dans les gènes
AGAMOUS ce qui rend leur détection impossible sur la base d’alignement de séquences. Le modèle biophysique surmonte ce type de difficulté et on peut espérer que son utilisation avec des séquences de plantes plus basales permettra de suivre les événements ayant conduit à l’apparition de la première fleur.
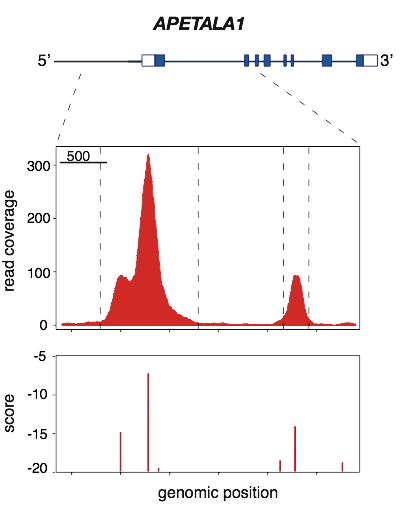
Figure : Exemple de correspondance entre la liaison de LEAFY à l’ADN
in vivo (représentée sur la partie haute de la figure par le nombre de lectures à chaque position du génome) et les prédictions de site de liaison issues du modèle construit in vitro (bas de la figure où les pics donnent le score du site prédit, proportionnel à l’affinité pour LFY du site correspondant).
Read coverage : nombre de lectures à chaque position du génome.
Genomic position : position sur le génome d'Arabidopsis thaliana.
Au-delà des questions d’évolution, ce type de modèle aidera à trouver, chez des espèces cultivées, les équivalents fonctionnels de régulateurs clés du développement ou de la réponse à l’environnement et leurs sites de fixation, de façon à pouvoir plus facilement transposer les résultats de recherche fondamentale vers des espèces d’intérêt agronomique.
ChIP-seq : technique utilisée pour analyser les interactions des protéines avec l'ADN in vivo. Le ChIP-Seq combine une immunoprécipitation de la chromatine (ChIP) et séquençage parallèle massif d'ADN pour identifier les régions du génome sur lesquelles des protéines s'associent. Il permet une cartographie précise des sites de liaison d'une protéine particulière.